Gene View
How are transcripts selected for the Gene View Page?¶
A single transcript was selected for determining the consequence types displayed for a gene based on the following rules:
- The transcript must be "protein coding" - if more than one transcript satisfies this rule then...
- The transcript must have an Ensembl "gencode basic" annotation flag - if more than one transcript satisfies this rule then...
- The transcript with an "CCDS" annotation flag - if more than one transcript satisfies this rule then...
- The transcript with the longest coding sequence length - if more than one transcript satisfies this rule then...
- The transcript with the largest number of exons is selected
If there is still more than one transcript for a gene then one is selected at random
How do I see a list of all loss of function variants in my gene?¶
From the "Variants" tab after performing a search you can filter variants using the "Consequence Type" filter
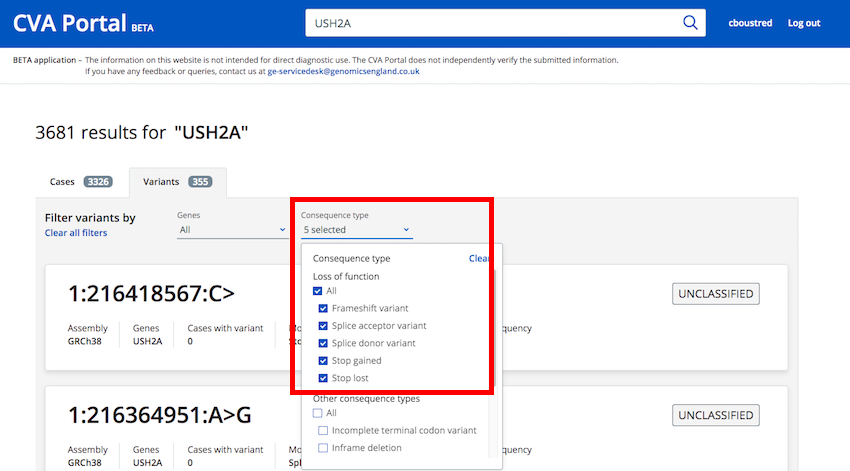
Loss of Function consequence types have been grouped into a single selection option
How do I see all classified variants in my gene?¶
The Gene Page can accessed here:
cva.genomicsengland.nhs.uk/gene/[HGNC symbol or ENSG]
e.g. https://cva.genomicsengland.nhs.uk/gene/APOE or https://cva.genomicsengland.nhs.uk/ENSG00000130203
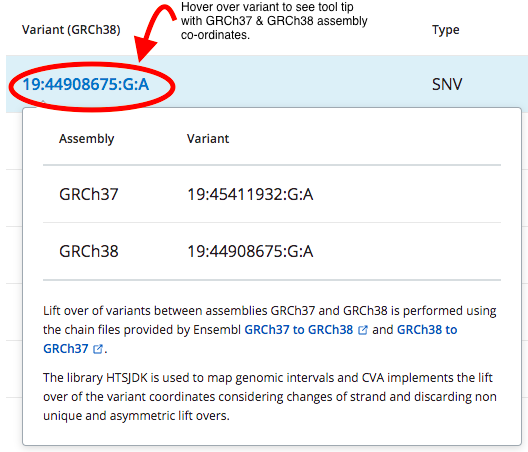
How do I know which genome assembly is relevant to my case?¶
All variants in the CVA database are lifted over to GRCh38, however, if you case was originally interpreted on GRCh37 these co-ordinates will be displayed.
You can "hover" over any variant co-ordinate in CVA Portal will show you the co-ordinates in both GRCh37 and GRCh38